
By Bushra Zletni, MSc
Receiving a life-changing diagnosis is always difficult, and for many families, a Huntington’s disease diagnosis introduces significant uncertainty and emotional burden. This condition progressively impacts movement, cognitive abilities, and mental well-being, influencing the everyday lives of both patients and their caregivers. For many years, the absence of treatments that modify the disease has rendered this diagnosis particularly tough; however, recent advancements indicate that a shift may finally be on the horizon.
Huntington’s disease is named after physician George Huntington, who first provided a clear clinical description of the condition in 1872. It still remains one of the most challenging neurodegenerative diseases to treat.
Contents
- What is Huntington’s disease?
- A new approach: one-time gene-based therapy
- Slowing disease progression
- Safety and side effects
- How long might the effect last?
- Limitations to this study
- Access, cost, and what comes next
- A cautious moment of hope
- Other references
DISCLAIMER: While I am a practising doctor, the information on this site is for educational purposes only. It does not take into account your personal circumstances, which can significantly affect medical decision-making and treatment. This content therefore does not constitute medical advice, and should not be relied upon for diagnosis or treatment. Always consult a qualified healthcare provider regarding any health concerns.
This article was written by a guest writer, and published on the 20/04/2026 using up-to-date sources at that time. Please be aware that medical information and guidelines change often.
What is Huntington’s disease?
Huntington’s disease is a progressive neurodegenerative condition that damages the brain over time. It occurs due to a change in the Huntingtin (HTT) gene — everyone has two copies of this gene, one inherited from each parent. In Huntington’s disease, one copy contains an expanded sequence of DNA (deoxyribonucleic acid) known as a CAG trinucleotide repeat. This trinucleotide codes for the amino acid* glutamine, so repeated sequences result in an abnormally long stretch of glutamine in the huntingtin protein, producing a toxic form of the protein.
*Amino acid = building blocks for all proteins.
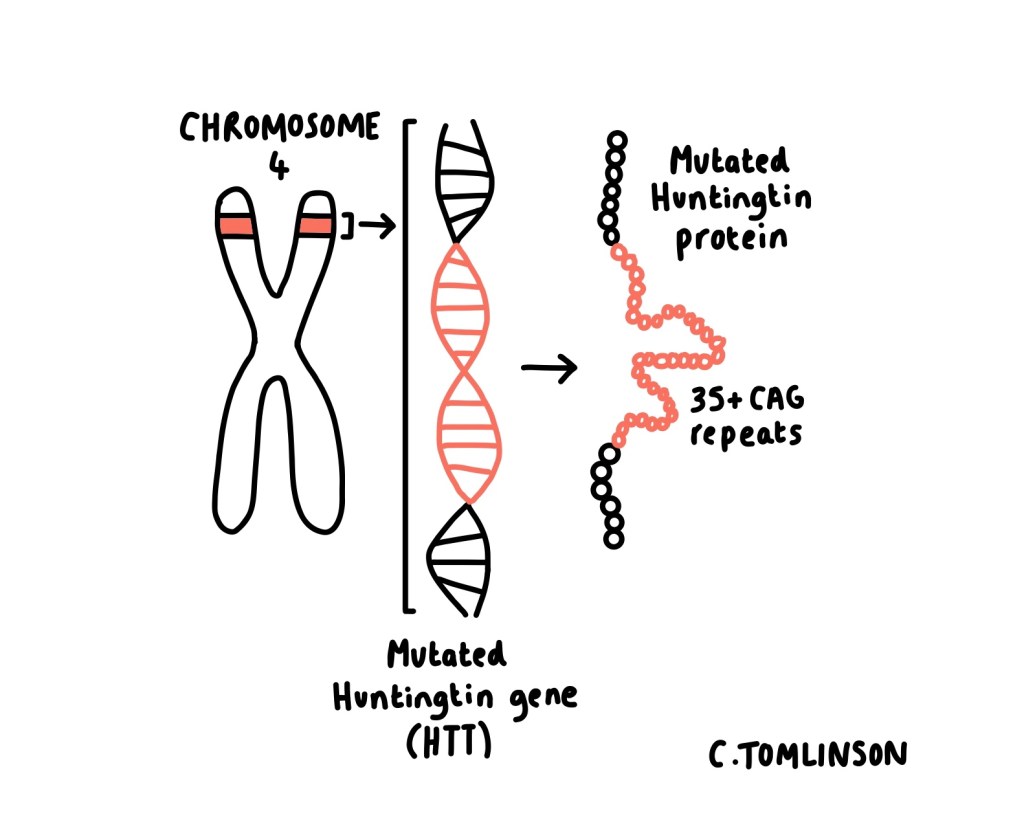
Editor’s note: Why the number of CAG repeats matters
The number of CAG repeats is crucial in determining whether the disease develops. Individuals with 28 or fewer repeats are unaffected. Those with 28–35 repeats do not develop the disease themselves but may pass on an expanded repeat to their children. 36–39 repeats are associated with reduced (variable) penetrance, meaning some individuals develop symptoms, often later in life. Individuals with 40 or more repeats will develop Huntington’s disease, with a greater number of repeats generally associated with earlier onset.
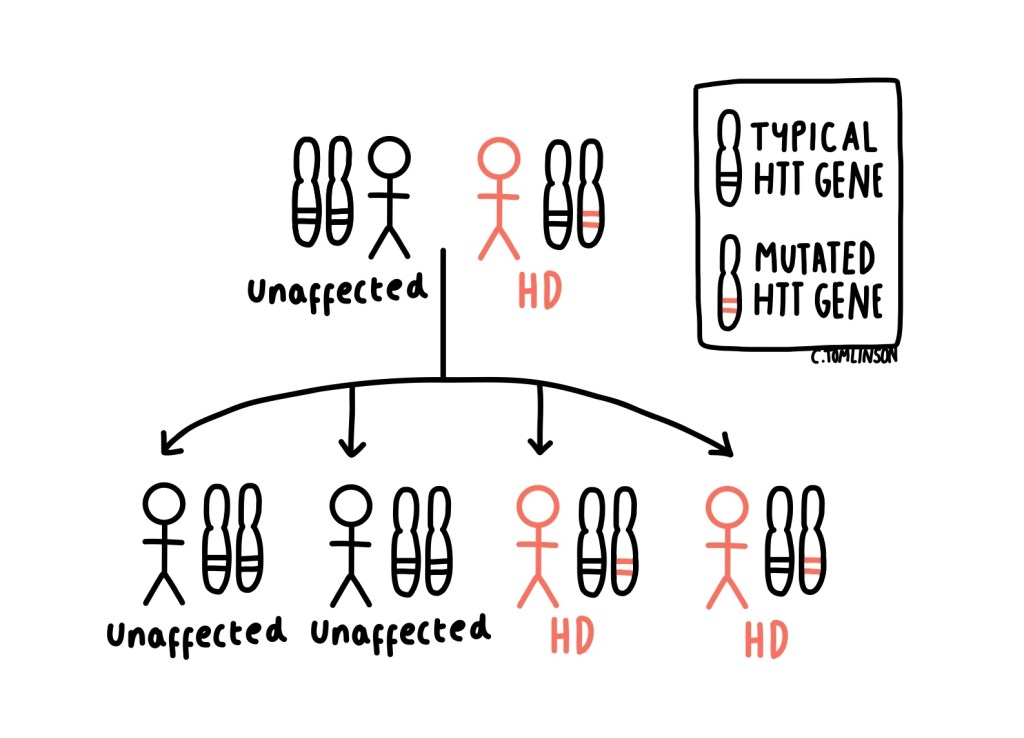
Over time, this toxic protein gradually damages nerve cells (neurones), particularly in areas of the brain involving movement, thinking, and emotional regulation. It is inherited in an autosomal dominant pattern, meaning each child has a 50% chance of inheriting the disease.
Symptoms most commonly present between the ages of 30 and 50 years and may include characteristic involuntary movements such as chorea (sudden, “dance-like” movements) and dystonia (sustained muscle contractions that can lead to twisting or abnormal postures), alongside progressive cognitive decline which may eventually lead to dementia, as well as behavioural or psychiatric symptoms. It is usually fatal within 15 to 20 years of symptom onset, with most deaths occurring as a result of complications of the disease, such as infections like pneumonia.
Huntington’s disease also shows a phenomenon known as genetic anticipation, where the number of CAG repeats can increase when passed to the next generation, often leading to earlier onset of symptoms in children compared with their affected parent.
Editor’s note: Current treatment options before gene therapy
Current treatment for Huntington’s disease focuses on managing symptoms rather than altering the course of the disease. Care is highly individualised and may include medications such as VMAT2 inhibitors (e.g. tetrabenazine) or antipsychotics to help control chorea and behavioural symptoms, alongside antidepressants for mood disorders. Physiotherapy, occupational therapy, and speech and language therapy play an important role in supporting mobility, communication, and daily functioning.
As the disease advances, care becomes increasingly supportive, with palliative care services involved to help manage symptoms and support patients and families in planning for future care.
A new approach: one-time gene-based therapy
Early results from a clinical trial of a one-time gene therapy called AMT‑130, developed by the biotechnology company uniQure and co-led in the UK by Professors Sarah Tabrizi and Ed Wild, suggest a promising new approach to treating Huntington’s disease.
In the UK part of the trial, 29 patients received the therapy, and their outcomes were compared with data from other patients who had not received the treatment. According to the company, the therapy is reported to slow disease progression by up to 75% over three years, meaning that symptoms developed more slowly than expected. These topline results were released by the company and reported widely in the news, but the full data have not yet appeared in peer-reviewed scientific publications, so experts continue to examine and interpret the findings.
This approach represents a novel combination of gene therapy and gene-silencing strategies, delivered during a single, highly specialised 12–18 hour neurosurgical procedure. Using real-time MRI scans, surgeons guide a small tube precisely to the regions of the brain most affected by Huntington’s disease: the caudate nucleus and the putamen. Through this tube, a harmless, modified virus carrying specially designed DNA is delivered directly into neurones. Once inside, the cells begin producing small genetic helpers known as microRNA. This microRNA acts like a traffic cop, blocking the signals that instruct the production of the toxic huntingtin protein. As a result, less harmful protein is produced in the brain, helping to slow disease progression.
Slowing disease progression
The disease progression was seen to be slowed down by 75 percent; this estimate was concluded through monitoring of learning and comprehension abilities, movement coordination, and the ability to perform daily tasks. In simpler terms, this treatment may significantly improve quality of life, where the decline in symptoms that would usually occur in one year would now take up to four years to develop.
Researchers also observed lower-than-expected levels of proteins that are usually released when neurones are damaged, suggesting a potential protective effect on brain cells. While participants’ identities were kept confidential, reports noted improvements in daily functioning, including greater mobility and independence in some individuals, with one individual reportedly being able to return to work.
Safety and side effects
Overall, the therapy was generally well-tolerated and safe. Some participants experienced side effects related to inflammation caused by the viral delivery system, including headaches and confusion. Most of these symptoms either resolved on their own or were managed with steroids.
How long might the effect last?
It is hoped that this treatment may have a long-lasting or even lifelong effect on patients with Huntington’s disease, mainly because it targets neurones which have a limited ability to regenerate. However, as this is a new treatment, doctors will continue to monitor patients over the coming years to confirm its long-term safety and effectiveness.
Limitations to this study
While the results are encouraging, several important limitations must be acknowledged, as they may affect how this treatment is applied in real-world clinical settings:
- The study was performed on a small number of participants. Small studies are more influenced by chance variations, such as one patient doing unusually well which could skew the results. In statistical terms, small sample sizes give less certainty about how well a treatment works, as they may show results that don’t run true in larger ones.
- The research has not been fully peer-reviewed nor published in peer-reviewed journals. Peer review is the process where independent experts check the methods, data, and conclusions before results are formally published in a scientific journal. Without peer review, there’s a higher risk that errors, missing details, or over interpretations haven’t yet been caught. This doesn’t mean that the results are wrong, but it means we should be cautious until the peer review process is complete.
- The surgery performed requires highly specialised skills and equipment.
- Long-term safety and effectiveness remain uncertain.
Access, cost, and what comes next
Naturally, gene therapies are expensive. A price tag hasn’t yet been put on this therapy, however, treatments in the same realm cost millions. This raises important questions about how widely the therapy could be made available through the NHS, and whether patients in need would be able to access it in a timely and equitable way.
According to UniQure, a regulatory approval application is expected in early 2026 in the United States, with discussions in the UK and Europe soon after. This is the formal process by which government health authorities review the safety and effectiveness of a new therapy before it can be prescribed to patients. If approved, this therapy could represent an important step toward slowing the progression of Huntington’s disease and potentially enabling earlier intervention.
A cautious moment of hope
For families affected by Huntington’s disease, these results provide a moment of cautious optimism. While this therapy is still under study, it seems to have the potential to slow symptom progression and help people maintain daily abilities and independence for longer. Researchers continue to monitor patients closely, and more work is needed to understand the full impact, but the findings offer a meaningful reason for hope.

Bushra Zletni holds an MSc in Advanced Biomedical Sciences from University College London (UCL) and a Bachelor’s degree in Biomedical Sciences from the University of Hull. She is an early-career medical writer and researcher with a strong interest in translating complex scientific concepts into clear, accessible content. She aspires to build a career in medical research and scientific writing, contributing to impactful work that advances understanding of human health and disease.
Other references:
- Gallagher J. Huntington’s disease successfully treated for first time. BBC News. 24 September 2025.
https://www.bbc.com/news/articles/cevz13xkxpro - Roos RAC. Huntington’s disease: a clinical review. Orphanet Journal of Rare Diseases. 2010;5:40.
https://pmc.ncbi.nlm.nih.gov/articles/PMC3022767/ - Caron NS, Wright GEB, Hayden MR. Huntington Disease. In: GeneReviews®. University of Washington, Seattle.
https://www.ncbi.nlm.nih.gov/books/NBK1305/ - Finkbeiner S. Huntington’s disease. Cold Spring Harbor Perspectives in Biology. 2011;3(6):a007476.
https://pubmed.ncbi.nlm.nih.gov/21576203/ - Tabrizi SJ, et al. Potential disease‑modifying therapies for Huntington’s disease. The Lancet Neurology. (Review article available in PubMed Central).
https://pubmed.ncbi.nlm.nih.gov/35716694/ - Pringsheim T, Wiltshire K, Day L, et al. The incidence and prevalence of Huntington’s disease: a systematic review and meta‑analysis.Movement Disorders. 2012;27(9):1083–1091. https://pubmed.ncbi.nlm.nih.gov/22692795/

Leave a comment